Pulmonary Hypertension Unit



What is Pulmonary Hypertension?
Pulmonary hypertension (PH) refers to high blood pressure in the blood vessels that supply the lungs (pulmonary= lungs; hypertension= high blood pressure). PH is classified into 5 different groups, depending on the underlying cause and predicted response to specific treatments. These include: (click/tap on table below for full size view)
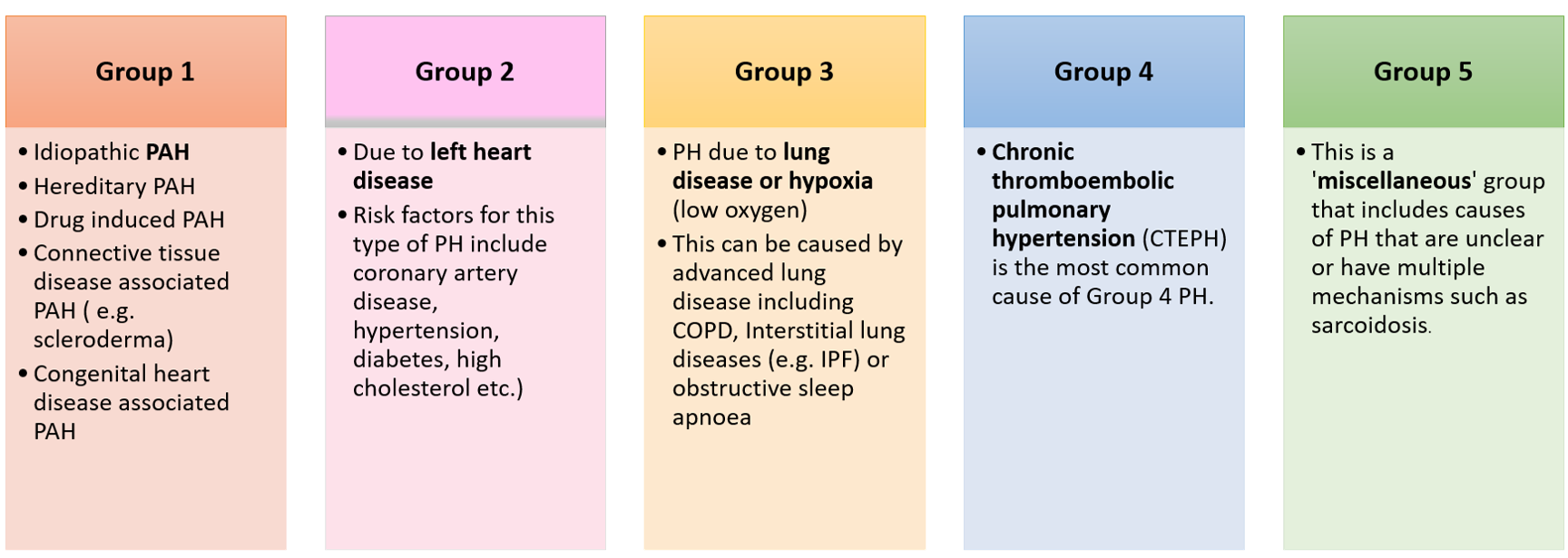
*PAH= Pulmonary arterial hypertension
Symptoms:
The diagnosis of PH is frequently delayed as the symptoms associated with this condition are often confused for other respiratory or cardiac conditions. These symptoms can include fatigue, shortness of breath, chest pain, palpitations, dizzy spells, faints and leg swelling. The diagnosis and subsequent classification of PH requires numerous investigations to differentiate between the various potential causes. More information regarding these investigations can be found here in our Investigation section.
The problem:
The heart is divided into right and left sided chambers. The role of the right heart is to push blood through the lungs, which is usually quite an easy thing to do. However, in pulmonary hypertension, these vessels are somewhat narrowed and the pressures in these vessels are higher than normal and therefore trying to pump blood from the right heart can be very difficult. If this is left untreated, the right heart is not able to cope with the extra workload and becomes exhausted, and this results in right heart failure. In order to avoid this we sometimes administer drugs called vasodilators, to open up these vessels and reduce the back pressure on the right heart.
Treatment:
Treatment decisions are guided by the underlying cause of the pulmonary hypertension. For example pulmonary arterial hypertension (PAH; Group 1) is typically treated with targeted PAH medications (More information available in the Medical Therapy section) and chronic thromboembolic PH (CTEPH; Group 4) may be amenable to specific surgical interventions. (More information is available in the Surgical Options section). The other groups of PH (Group 2, 3 and 5) are often best served by treating the underlying condition that has caused the PH, as targeted PAH medication may be unlikely to provide beneficial effects and may even cause harm in certain instances. Therefore, treatment decisions in clinic are always made on an individual, case-by-case basis and guided by your symptoms, functional ability and treatment preferences.
